
Genomics — the study of the genome — requires processing and analysis of large-scale datasets. For example, to identify genes that increase our susceptibility to a particular disease, scientists can read many genomes of individuals with and without this disease and try to compare differences among genomes. After running human samples on DNA sequencers, scientists need to work with overwhelming datasets.
Working with genomic datasets requires knowledge of a myriad of bioinformatic software packages. Because of rapid development of bioinformatic tools, it can be difficult to decide which software packages to use. For example, there are at least 18 software packages to align short unspliced DNA reads to a reference genome. Of course, scientists make this choice routinely and their collective knowledge is embedded in the literature.
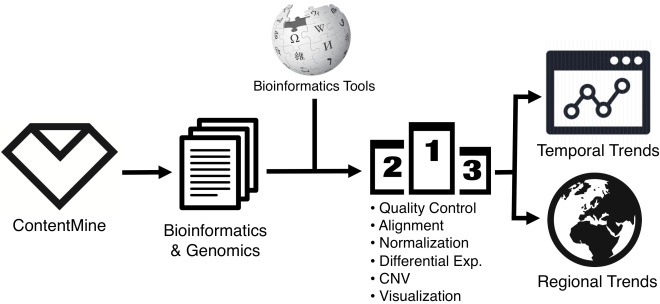
To this end, I have been using ContentMine to find trends in bioinformatic tools. This blog post outlines my approach and demonstrates important challenges to address. Much of my current efforts are in obtaining as comprehensive a set of articles as possible, ensuring correct counting/matching methods for bioinformatic packages, and making this pipeline reproducible.
Continue reading Evaluating Trends in Bioinformatics Software Packages











