
Lars Willighagen, orcid:0000-0002-4751-4637
Final Report of my fellowship at the ContentMine.
Proposal
My proposal was to extract facts about various conifer species by analysing text from papers with software suited for analysing text and the tools provided by the ContentMine. These facts were then to be converted into JSON, and then viewable with an HTML (+CSS/JS) interface. Expected statements were like: ‘Picea glauca is a species of the genus Picea’, which could be parsed to the triple:Picea glauca; property:genus; subject:Picea.
Work
The main outcome of this project is a series of programmes converting tables from research articles into Wikidata statements. The workflow is as follows. First, papers matching a user-provided query are fetched by the ContentMine’s getpapers. Second, the tables are extracted from the fetched papers and converted to assertions. This is done by filling empty cells in tables and then treating each row as an object, the first column being the name and the others property-value pairs. Different table designs are currently parsed in the same way, resulting in incorrect extraction of data, something that can be accommodated for by normalising the table structure beforehand. The resulting assertions are then converted to JSON, currently in a custom scheme, to allow the next steps.
Finally, the JSON assertions are visualized in an HTML GUI. This includes a stepper form (see picture) where you can curate the assertion, link identifiers, and add it to Wikidata.

- Source code: https://github.com/larsgw/ctj-factvis
- Demo: https://larsgw.github.io/ctj-factvis
Getting these assertions from text, as I proposed, was harder. Tools I expected to find included in ContentMine software were nowhere to be found, but were planned, so actually implementing them myself did not seem a good use of my time. Luckily, the literature corpus does not actually contain that many statements about physical properties of conifers in plain text as I originally expected: most are in tables, figures or in supplementary files, leading me to using those instead. The nice thing is that one of the main focuses of the ContentMine is parsing tables from PDF, so this will definitely be of general use.
Other work
During the project and to explore the design of the ContentMine, additional related components were developed:
- ctj: program to convert and re-order AMI data to JSON, making it easier to read in JavaScript (mainly good for web applications);
- ctj-cardlists: program to view AMI JSON (see above) in a Web GUI (demo); and
- Citation.js: added functionality to parse BibJSON (used for quickscrape output) into CSL, for further formatting. See blog post.
These first two simplified handing AMI output in the browser, while the third makes it easier to display references in common formats.
Dissemination
All source code of the project outcomes is available on GitHub:
Progress was communicated during the project via the ContentMine Discourse page, on my personal blog (~20 posts), and on the general ContentMining blog (2 long posts).
Future work
The developed pipeline works but is not perfect.The pipeline to parse tables mentioned above requires further generalisation. This defines some logical next steps: fixes:
- Finally adding it as an NPM module, making it (way) easier for people to use it;
- Making searching easier in the HTML GUI (will need work further upstream too). Currently the list of assertions are split into pieces, making it hard to find anything. This can be fixed with a search index;
- Normalising table structures to support more designs, rendering assertion extraction more reliable;
- Making the process of curating assertions and linking identifiers easier by linking more identifiers, and showing context, i.e. the original tables; and
- Some small performance and UX things.
Another important thing that is too big for a single bullet point, is annotating abbreviations and references in the document before extracting the tables. It’s easier to curate statements like ‘[1] says this and this’ when you know ‘[1]’ references some known article. Another example: while a statement containing ‘P. glauca’ says nothing (there are 66+ species using that abbreviation), the article probably says which one it is somewhere outside the table, something that can be picked up if you annotate these before taking them out of context. This makes the interactive stepper form currently a necessity.
Evaluation
As noted, the work is far from done. Currently, it mainly shows a glimpse of what is possible had I spent more time on writing code. Short conclusions: CTJ is unpolished and slow. Because of a lack of customisation options, such as what data to use, you will almost always need to write custom code to not have to include tons of unnecessary data in your resulting JSON.
CTJ-Cardlists is actually pretty nice. It is slow, and it does not really show relations, but it does show an interesting overview of the literature corpus, like how often species are mentioned and with what they are mentioned together most of the time. You can easily draw reasonable conclusions like how often species names are misspelled. However, it would be more useful for this to have SQL queries or something similar. CTJ-Factvis shows even more potential, with the Wikidata integration. I do need to pay more attention to the fact that those assertions are alleged facts, and not regular ones, as I called them in earlier blog posts.
Fellowship
In general, the fellowship went pretty well for me. In retrospect, I did a lot of the things I wanted to do, even though that throughout the project it felt like there was so much left to do, and there is! I am really excited about the possibilities that emerged during the fellowship, and even in the last weeks. How cool would it be to extend this project with entire Web API’s and more? This is, for a big part, thanks to the support, feedback, and input of the amazing ContentMine team during the regular meeting, and the quick responses to various software issues. I also enjoyed blogging about my progress on my own blog and on the ContentMine blog.


 I am a taxonomist, the kind of biologists who are charged with discovering, documenting and describing life on earth. I specialize on insects, the most diverse and successful form of life. My ContentMine Fellowship project will focus on mining weevil-plant associations from literature records. I will describe my project in the following.
I am a taxonomist, the kind of biologists who are charged with discovering, documenting and describing life on earth. I specialize on insects, the most diverse and successful form of life. My ContentMine Fellowship project will focus on mining weevil-plant associations from literature records. I will describe my project in the following.

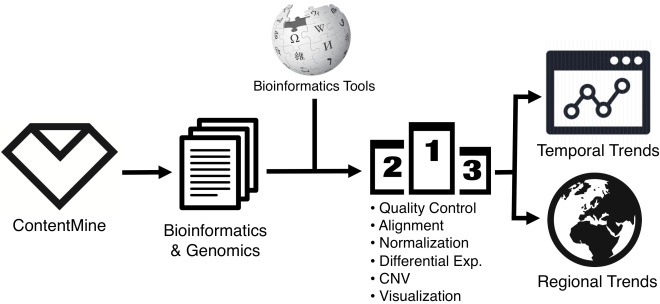
 High-throughput genomics has fundamentally changed how life scientists investigate their research questions. Instead of studying a single candidate gene, we are able to measure DNA sequences and RNA expression of every gene in a large number of genomes. This technology advance requires us to become familiar with data wrangling, programming, and statistical analysis. To meet new challenges, bioinformaticians have created a large number of software packages to deal with genomic data.
High-throughput genomics has fundamentally changed how life scientists investigate their research questions. Instead of studying a single candidate gene, we are able to measure DNA sequences and RNA expression of every gene in a large number of genomes. This technology advance requires us to become familiar with data wrangling, programming, and statistical analysis. To meet new challenges, bioinformaticians have created a large number of software packages to deal with genomic data.